

Target Information
| Target General Information | Top | |||||
|---|---|---|---|---|---|---|
| Target ID |
T68698
(Former ID: TTDR00957)
|
|||||
| Target Name |
Adenosylhomocysteinase (AHCY)
|
|||||
| Synonyms |
SAHH; SAH hydrolase; S-adenosyl-L-homocysteine hydrolase
Click to Show/Hide
|
|||||
| Gene Name |
AHCY
|
|||||
| Target Type |
Literature-reported target
|
[1] | ||||
| Function |
Adenosylhomocysteine is a competitive inhibitor of S-adenosyl-L-methionine-dependent methyl transferase reactions; therefore adenosylhomocysteinase may play a key role in the control of methylations via regulation of the intracellular concentration of adenosylhomocysteine.
Click to Show/Hide
|
|||||
| BioChemical Class |
Ether bond hydrolase
|
|||||
| UniProt ID | ||||||
| EC Number |
EC 3.3.1.1
|
|||||
| Sequence |
MSDKLPYKVADIGLAAWGRKALDIAENEMPGLMRMRERYSASKPLKGARIAGCLHMTVET
AVLIETLVTLGAEVQWSSCNIFSTQDHAAAAIAKAGIPVYAWKGETDEEYLWCIEQTLYF KDGPLNMILDDGGDLTNLIHTKYPQLLPGIRGISEETTTGVHNLYKMMANGILKVPAINV NDSVTKSKFDNLYGCRESLIDGIKRATDVMIAGKVAVVAGYGDVGKGCAQALRGFGARVI ITEIDPINALQAAMEGYEVTTMDEACQEGNIFVTTTGCIDIILGRHFEQMKDDAIVCNIG HFDVEIDVKWLNENAVEKVNIKPQVDRYRLKNGRRIILLAEGRLVNLGCAMGHPSFVMSN SFTNQVMAQIELWTHPDKYPVGVHFLPKKLDEAVAEAHLGKLNVKLTKLTEKQAQYLGMS CDGPFKPDHYRY Click to Show/Hide
|
|||||
| 3D Structure | Click to Show 3D Structure of This Target | PDB | ||||
| HIT2.0 ID | T94T7V | |||||
| Cell-based Target Expression Variations | Top | |||||
|---|---|---|---|---|---|---|
| Cell-based Target Expression Variations | ||||||
| Drug Binding Sites of Target | Top | |||||
|---|---|---|---|---|---|---|
| Ligand Name: Adenosine | Ligand Info | |||||
| Structure Description | The structure of bi-acetylated SAHH | PDB:4PFJ | ||||
| Method | X-ray diffraction | Resolution | 2.30 Å | Mutation | Yes | [11] |
| PDB Sequence |
DKLPYKVADI
12 GLAAWGRKAL22 DIAENEMPGL32 MRMRERYSAS42 KPLKGARIAG52 CLHMTVETAV 62 LIETLVTLGA72 EVQWSSCNIF82 STQNHAAAAI92 AKAGIPVYAW102 KGETDEEYLW 112 CIEQTLYFKD122 GPLNMILDDG132 GDLTNLIHTK142 YPQLLPGIRG152 ISEETTTGVH 162 NLYKMMANGI172 LKVPAINVND182 SVTKSKFDNL192 YGCRESLIDG202 IKRATDVMIA 212 GKVAVVAGYG222 DVGKGCAQAL232 RGFGARVIIT242 EIDPINALQA252 AMEGYEVTTM 262 DEACQEGNIF272 VTTTGCIDII282 LGRHFEQMKD292 DAIVCNIGHF302 DVEIDVKWLN 312 ENAVEKVNIK322 PQVDRYRLKN332 GRRIILLAEG342 RLVNLGCAMG352 HPSFVMSNSF 362 TNQVMAQIEL372 WTHPDKYPVG382 VHFLPKKLDE392 AVACAHLGLN403 VKLTLTEKQA 414 QYLGMSCDGP424 FKPDHYRY
|
|||||
|
|
LEU54
2.577
HIS55
2.766
THR57
2.652
GLU59
2.775
THR60
2.318
CYS79
4.328
GLN85
3.556
ASP131
2.478
GLU156
2.575
THR157
2.116
THR158
3.898
ASN181
4.239
LYS186
1.922
ASP190
2.123
ASN191
4.721
|
|||||
| Click to View More Binding Site Information of This Target and Ligand Pair | ||||||
| Ligand Name: NADH | Ligand Info | |||||
| Structure Description | Structure of S-adenosyl-L-homocysteine hydrolase | PDB:4YVF | ||||
| Method | X-ray diffraction | Resolution | 2.70 Å | Mutation | No | [12] |
| PDB Sequence |
KLPYKVADIG
13 LAAWGRKALD23 IAENEMPGLM33 RMRERYSASK43 PLKGARIAGC53 LHMTVETAVL 63 IETLVTLGAE73 VQWSSCNIFS83 TQDHAAAAIA93 KAGIPVYAWK103 GETDEEYLWC 113 IEQTLYFKDG123 PLNMILDDGG133 DLTNLIHTKY143 PQLLPGIRGI153 SEETTTGVHN 163 LYKMMANGIL173 KVPAINVNDS183 VTKSKFDNLY193 GCRESLIDGI203 KRATDVMIAG 213 KVAVVAGYGD223 VGKGCAQALR233 GFGARVIITE243 IDPINALQAA253 MEGYEVTTMD 263 EACQEGNIFV273 TTTGCIDIIL283 GRHFEQMKDD293 AIVCNIGHFD303 VEIDVKWLNE 313 NAVEKVNIKP323 QVDRYRLKNG333 RRIILLAEGR343 LVNLGCAMGH353 PSFVMSNSFT 363 NQVMAQIELW373 THPDKYPVGV383 HFLPKKLDEA393 VAEAHLGKLN403 VKLTKLTEKQ 413 AQYLGMSCDG423 PFKPDHYRY
|
|||||
|
|
THR158
4.813
ASP190
3.108
ASN191
3.109
CYS195
3.549
ALA219
4.618
GLY220
3.802
TYR221
4.341
GLY222
3.328
ASP223
3.214
VAL224
2.804
GLY225
4.201
THR242
3.430
GLU243
2.728
ILE244
3.376
|
|||||
| Click to View More Binding Site Information of This Target with Different Ligands | ||||||
| Different Human System Profiles of Target | Top |
|---|---|
|
Human Similarity Proteins
of target is determined by comparing the sequence similarity of all human proteins with the target based on BLAST. The similarity proteins for a target are defined as the proteins with E-value < 0.005 and outside the protein families of the target.
A target that has fewer human similarity proteins outside its family is commonly regarded to possess a greater capacity to avoid undesired interactions and thus increase the possibility of finding successful drugs
(Brief Bioinform, 21: 649-662, 2020).
Human Tissue Distribution
of target is determined from a proteomics study that quantified more than 12,000 genes across 32 normal human tissues. Tissue Specificity (TS) score was used to define the enrichment of target across tissues.
The distribution of targets among different tissues or organs need to be taken into consideration when assessing the target druggability, as it is generally accepted that the wider the target distribution, the greater the concern over potential adverse effects
(Nat Rev Drug Discov, 20: 64-81, 2021).
Human Pathway Affiliation
of target is determined by the life-essential pathways provided on KEGG database. The target-affiliated pathways were defined based on the following two criteria (a) the pathways of the studied target should be life-essential for both healthy individuals and patients, and (b) the studied target should occupy an upstream position in the pathways and therefore had the ability to regulate biological function.
Targets involved in a fewer pathways have greater likelihood to be successfully developed, while those associated with more human pathways increase the chance of undesirable interferences with other human processes
(Pharmacol Rev, 58: 259-279, 2006).
Biological Network Descriptors
of target is determined based on a human protein-protein interactions (PPI) network consisting of 9,309 proteins and 52,713 PPIs, which were with a high confidence score of ≥ 0.95 collected from STRING database.
The network properties of targets based on protein-protein interactions (PPIs) have been widely adopted for the assessment of target’s druggability. Proteins with high node degree tend to have a high impact on network function through multiple interactions, while proteins with high betweenness centrality are regarded to be central for communication in interaction networks and regulate the flow of signaling information
(Front Pharmacol, 9, 1245, 2018;
Curr Opin Struct Biol. 44:134-142, 2017).
Human Similarity Proteins
Human Tissue Distribution
Human Pathway Affiliation
Biological Network Descriptors
|
|
|
There is no similarity protein (E value < 0.005) for this target
|
|
Note:
If a protein has TS (tissue specficity) scores at least in one tissue >= 2.5, this protein is called tissue-enriched (including tissue-enriched-but-not-specific and tissue-specific). In the plots, the vertical lines are at thresholds 2.5 and 4.
|
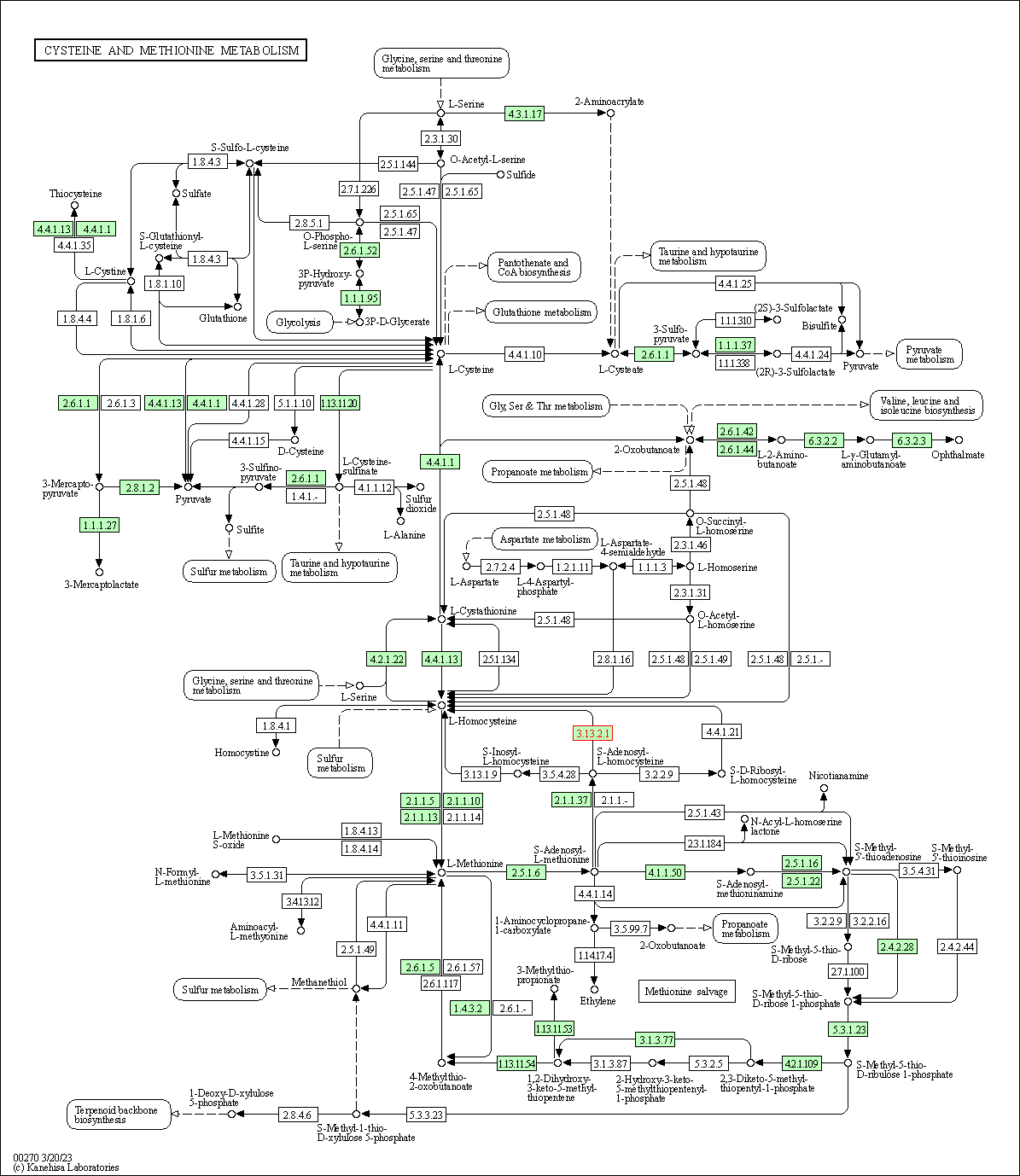
| KEGG Pathway | Pathway ID | Affiliated Target | Pathway Map |
|---|---|---|---|
| Cysteine and methionine metabolism | hsa00270 | Affiliated Target |
|
| Class: Metabolism => Amino acid metabolism | Pathway Hierarchy | ||
| Degree | 6 | Degree centrality | 6.45E-04 | Betweenness centrality | 2.17E-03 |
|---|---|---|---|---|---|
| Closeness centrality | 2.02E-01 | Radiality | 1.35E+01 | Clustering coefficient | 2.00E-01 |
| Neighborhood connectivity | 1.38E+01 | Topological coefficient | 1.96E-01 | Eccentricity | 12 |
| Download | Click to Download the Full PPI Network of This Target | ||||
| Chemical Structure based Activity Landscape of Target | Top |
|---|---|
| Drug Property Profile of Target | Top | |
|---|---|---|
| (1) Molecular Weight (mw) based Drug Clustering | (2) Octanol/Water Partition Coefficient (xlogp) based Drug Clustering | |
|
|
||
| (3) Hydrogen Bond Donor Count (hbonddonor) based Drug Clustering | (4) Hydrogen Bond Acceptor Count (hbondacc) based Drug Clustering | |
|
|
||
| (5) Rotatable Bond Count (rotbonds) based Drug Clustering | (6) Topological Polar Surface Area (polararea) based Drug Clustering | |
|
|
||
| "RO5" indicates the cutoff set by lipinski's rule of five; "D123AB" colored in GREEN denotes the no violation of any cutoff in lipinski's rule of five; "D123AB" colored in PURPLE refers to the violation of only one cutoff in lipinski's rule of five; "D123AB" colored in BLACK represents the violation of more than one cutoffs in lipinski's rule of five | ||
| Target Poor or Non Binders | Top | |||||
|---|---|---|---|---|---|---|
| Target Poor or Non Binders | ||||||
| Target Regulators | Top | |||||
|---|---|---|---|---|---|---|
| Target-interacting Proteins | ||||||
| Target Affiliated Biological Pathways | Top | |||||
|---|---|---|---|---|---|---|
| BioCyc | [+] 3 BioCyc Pathways | + | ||||
| 1 | Superpathway of methionine degradation | |||||
| 2 | Methionine degradation | |||||
| 3 | Cysteine biosynthesis | |||||
| KEGG Pathway | [+] 2 KEGG Pathways | + | ||||
| 1 | Cysteine and methionine metabolism | |||||
| 2 | Metabolic pathways | |||||
| Pathwhiz Pathway | [+] 3 Pathwhiz Pathways | + | ||||
| 1 | Selenoamino Acid Metabolism | |||||
| 2 | Betaine Metabolism | |||||
| 3 | Methionine Metabolism | |||||
| WikiPathways | [+] 6 WikiPathways | + | ||||
| 1 | Metabolism of amino acids and derivatives | |||||
| 2 | Trans-sulfuration and one carbon metabolism | |||||
| 3 | One Carbon Metabolism | |||||
| 4 | Trans-sulfuration pathway | |||||
| 5 | Phase II conjugation | |||||
| 6 | Folate Metabolism | |||||
| Target-Related Models and Studies | Top | |||||
|---|---|---|---|---|---|---|
| Target Validation | ||||||
| References | Top | |||||
|---|---|---|---|---|---|---|
| REF 1 | Molecular approaches for the treatment of hemorrhagic fever virus infections. Antiviral Res. 1993 Sep;22(1):45-75. | |||||
| REF 2 | Synthesis of 2-modified aristeromycins and their analogs as potent inhibitors against Plasmodium falciparum S-adenosyl-L-homocysteine hydrolase. Bioorg Med Chem. 2008 Apr 1;16(7):3809-15. | |||||
| REF 3 | The Protein Data Bank. Nucleic Acids Res. 2000 Jan 1;28(1):235-42. | |||||
| REF 4 | DrugBank 3.0: a comprehensive resource for 'omics' research on drugs. Nucleic Acids Res. 2011 Jan;39(Database issue):D1035-41. | |||||
| REF 5 | Synthesis of 5'-functionalized nucleosides: S-Adenosylhomocysteine analogues with the carbon-5' and sulfur atoms replaced by a vinyl or halovinyl u... Bioorg Med Chem. 2008 May 15;16(10):5424-33. | |||||
| REF 6 | Design, synthesis, and molecular modeling studies of 5'-deoxy-5'-ureidoadenosine: 5'-ureido group as multiple hydrogen bonding donor in the active ... Bioorg Med Chem Lett. 2007 Aug 15;17(16):4456-9. | |||||
| REF 7 | How many drug targets are there Nat Rev Drug Discov. 2006 Dec;5(12):993-6. | |||||
| REF 8 | 3-Deazaneplanocin: a new and potent inhibitor of S-adenosylhomocysteine hydrolase and its effects on human promyelocytic leukemia cell line HL-60. Biochem Biophys Res Commun. 1986 Mar 13;135(2):688-94. | |||||
| REF 9 | Synthesis of 5'-substituted fluoro-neplanocin A analogues: importance of a hydrogen bonding donor at 5'-position for the inhibitory activity of S-a... Bioorg Med Chem Lett. 2004 Nov 15;14(22):5641-4. | |||||
| REF 10 | Synthesis of 4'-modified noraristeromycins to clarify the effect of the 4'-hydroxyl groups for inhibitory activity against S-adenosyl-L-homocystein... Bioorg Med Chem Lett. 2008 Apr 15;18(8):2615-8. | |||||
| REF 11 | Regulation of S-adenosylhomocysteine hydrolase by lysine acetylation. J Biol Chem. 2014 Nov 7;289(45):31361-72. | |||||
| REF 12 | Discovery and structural analyses of S-adenosyl-L-homocysteine hydrolase inhibitors based on non-adenosine analogs. Bioorg Med Chem. 2015 Aug 1;23(15):4952-4969. | |||||
If You Find Any Error in Data or Bug in Web Service, Please Kindly Report It to Dr. Zhou and Dr. Zhang.