

Target Information
| Target General Information | Top | |||||
|---|---|---|---|---|---|---|
| Target ID |
T52450
(Former ID: TTDC00147)
|
|||||
| Target Name |
Matrix metalloproteinase-1 (MMP-1)
|
|||||
| Synonyms |
Interstitial collagenase; Fibroblast collagenase; CLG
Click to Show/Hide
|
|||||
| Gene Name |
MMP1
|
|||||
| Target Type |
Successful target
|
[1] | ||||
| Disease | [+] 1 Target-related Diseases | + | ||||
| 1 | Lung cancer [ICD-11: 2C25] | |||||
| Function |
Cleaves collagens of types VII and X. In case of HIV infection, interacts and cleaves the secreted viral Tat protein, leading to a decrease in neuronal Tat's mediated neurotoxicity. Cleaves collagens of types I, II, and III at one site in the helical domain.
Click to Show/Hide
|
|||||
| BioChemical Class |
Peptidase
|
|||||
| UniProt ID | ||||||
| EC Number |
EC 3.4.24.7
|
|||||
| Sequence |
MHSFPPLLLLLFWGVVSHSFPATLETQEQDVDLVQKYLEKYYNLKNDGRQVEKRRNSGPV
VEKLKQMQEFFGLKVTGKPDAETLKVMKQPRCGVPDVAQFVLTEGNPRWEQTHLTYRIEN YTPDLPRADVDHAIEKAFQLWSNVTPLTFTKVSEGQADIMISFVRGDHRDNSPFDGPGGN LAHAFQPGPGIGGDAHFDEDERWTNNFREYNLHRVAAHELGHSLGLSHSTDIGALMYPSY TFSGDVQLAQDDIDGIQAIYGRSQNPVQPIGPQTPKACDSKLTFDAITTIRGEVMFFKDR FYMRTNPFYPEVELNFISVFWPQLPNGLEAAYEFADRDEVRFFKGNKYWAVQGQNVLHGY PKDIYSSFGFPRTVKHIDAALSEENTGKTYFFVANKYWRYDEYKRSMDPGYPKMIAHDFP GIGHKVDAVFMKDGFFYFFHGTRQYKFDPKTKRILTLQKANSWFNCRKN Click to Show/Hide
|
|||||
| 3D Structure | Click to Show 3D Structure of This Target | PDB | ||||
| HIT2.0 ID | T80OJF | |||||
| Drugs and Modes of Action | Top | |||||
|---|---|---|---|---|---|---|
| Approved Drug(s) | [+] 1 Approved Drugs | + | ||||
| 1 | Prinomastat | Drug Info | Approved | Lung cancer | [2], [3] | |
| Clinical Trial Drug(s) | [+] 3 Clinical Trial Drugs | + | ||||
| 1 | CIPEMASTAT | Drug Info | Phase 3 | Rheumatoid arthritis | [4], [5] | |
| 2 | Marimastat | Drug Info | Phase 3 | Pancreatic cancer | [6], [7] | |
| 3 | Apratastat | Drug Info | Phase 2 | Rheumatoid arthritis | [8], [9] | |
| Discontinued Drug(s) | [+] 8 Discontinued Drugs | + | ||||
| 1 | BMS 275291 | Drug Info | Discontinued in Phase 3 | Kaposi sarcoma | [10] | |
| 2 | GM6001 | Drug Info | Discontinued in Phase 2 | Corneal ulcer | [11] | |
| 3 | RS-130830 | Drug Info | Discontinued in Phase 2 | Hepatitis C virus infection | [12] | |
| 4 | XL784 | Drug Info | Discontinued in Phase 2 | Diabetic nephropathy | [13] | |
| 5 | BB-1101 | Drug Info | Terminated | Multiple sclerosis | [14] | |
| 6 | BB-3644 | Drug Info | Terminated | Solid tumour/cancer | [15] | |
| 7 | Ro-31-4724 | Drug Info | Terminated | Arthritis | [16] | |
| 8 | RO-319790 | Drug Info | Terminated | Rheumatoid arthritis | [17] | |
| Mode of Action | [+] 2 Modes of Action | + | ||||
| Inhibitor | [+] 37 Inhibitor drugs | + | ||||
| 1 | Prinomastat | Drug Info | [1], [18], [19], [20], [21] | |||
| 2 | Marimastat | Drug Info | [22], [23], [24], [21] | |||
| 3 | Apratastat | Drug Info | [25] | |||
| 4 | PMID29130358-Compound-Figure10(2a) | Drug Info | [26] | |||
| 5 | PMID29130358-Compound-Figure18(14a) | Drug Info | [26] | |||
| 6 | BMS 275291 | Drug Info | [10], [27], [28], [21] | |||
| 7 | GM6001 | Drug Info | [29] | |||
| 8 | RS-130830 | Drug Info | [30] | |||
| 9 | XL784 | Drug Info | [13] | |||
| 10 | BB-1101 | Drug Info | [31] | |||
| 11 | BB-3644 | Drug Info | [21], [15] | |||
| 12 | L-696418 | Drug Info | [32] | |||
| 13 | Ro-31-4724 | Drug Info | [33] | |||
| 14 | RO-319790 | Drug Info | [34] | |||
| 15 | SC-44463 | Drug Info | [35] | |||
| 16 | 3-(4-Methoxy-benzenesulfonyl)-cyclohexanethiol | Drug Info | [36] | |||
| 17 | 3-(4-Methoxy-benzenesulfonyl)-hexane-1-thiol | Drug Info | [37] | |||
| 18 | 3-(4-Methoxy-benzenesulfonyl)-pentane-1-thiol | Drug Info | [37] | |||
| 19 | 3-(4-Phenoxy-benzenesulfonyl)-cyclohexanethiol | Drug Info | [36] | |||
| 20 | 3-(4-Phenoxy-benzenesulfonyl)-propane-1-thiol | Drug Info | [37] | |||
| 21 | 3-Benzenesulfinyl-heptanoic acid hydroxyamide | Drug Info | [38] | |||
| 22 | 3-Benzenesulfonyl-heptanoic acid hydroxyamide | Drug Info | [38] | |||
| 23 | 3-Cyclohexanesulfonyl-heptanoic acid hydroxyamide | Drug Info | [38] | |||
| 24 | 4-(4-Butoxy-phenyl)-N-hydroxy-4-oxo-butyramide | Drug Info | [39] | |||
| 25 | 4-(4-Methoxy-benzenesulfonyl)-butane-2-thiol | Drug Info | [37] | |||
| 26 | 4-Butoxy-N-hydroxycarbamoylmethyl-benzamide | Drug Info | [39] | |||
| 27 | METHYLAMINO-PHENYLALANYL-LEUCYL-HYDROXAMIC ACID | Drug Info | [40] | |||
| 28 | MMI270 | Drug Info | [41] | |||
| 29 | N-(Ethylphosphoryl)-L-isoleucyl-L-Trp-NHCH3 | Drug Info | [42] | |||
| 30 | N-Hydroxy-4-(4-methoxy-phenyl)-4-oxo-butyramide | Drug Info | [39] | |||
| 31 | N-Hydroxy-4-oxo-4-(4-phenoxy-phenyl)-butyramide | Drug Info | [39] | |||
| 32 | N-Hydroxycarbamoylmethyl-4-methoxy-benzamide | Drug Info | [39] | |||
| 33 | N-Hydroxycarbamoylmethyl-4-phenoxy-benzamide | Drug Info | [39] | |||
| 34 | PKF-242-484 | Drug Info | [43] | |||
| 35 | Ro-37-9790 | Drug Info | [44] | |||
| 36 | RS-39066 | Drug Info | [45] | |||
| 37 | SR-973 | Drug Info | [46] | |||
| Modulator | [+] 1 Modulator drugs | + | ||||
| 1 | CIPEMASTAT | Drug Info | [5] | |||
| Cell-based Target Expression Variations | Top | |||||
|---|---|---|---|---|---|---|
| Cell-based Target Expression Variations | ||||||
| Drug Binding Sites of Target | Top | |||||
|---|---|---|---|---|---|---|
| Ligand Name: MMI270 | Ligand Info | |||||
| Structure Description | CATALYTIC FRAGMENT OF HUMAN FIBROBLAST COLLAGENASE COMPLEXED WITH CGS-27023A, NMR, MINIMIZED AVERAGE STRUCTURE | PDB:3AYK | ||||
| Method | Solution NMR | Resolution | N.A. | Mutation | No | [47] |
| PDB Sequence |
PRWEQTHLTY
16 RIENYTPDLP26 RADVDHAIEK36 AFQLWSNVTP46 LTFTKVSEGQ56 ADIMISFVRG 66 DHRDNSPFDG76 PGGNLAHAFQ86 PGPGIGGDAH96 FDEDERWTNN106 FREYNLHRVA 116 AHELGHSLGL126 SHSTDIGALM136 YPSYTFSGDV146 QLAQDDIDGI156 QAIYGRS |
|||||
|
|
||||||
| Click to View More Binding Site Information of This Target and Ligand Pair | ||||||
| Ligand Name: METHYLAMINO-PHENYLALANYL-LEUCYL-HYDROXAMIC ACID | Ligand Info | |||||
| Structure Description | 1.56 ANGSTROM STRUCTURE OF MATURE TRUNCATED HUMAN FIBROBLAST COLLAGENASE | PDB:1HFC | ||||
| Method | X-ray diffraction | Resolution | 1.50 Å | Mutation | No | [48] |
| PDB Sequence |
PRWEQTHLTY
116 RIENYTPDLP126 RADVDHAIEK136 AFQLWSNVTP146 LTFTKVSEGQ156 ADIMISFVRG 166 DHRDNSPFDG176 PGGNLAHAFQ186 PGPGIGGDAH196 FDEDERWTNN206 FREYNLHRVA 216 AHELGHSLGL226 SHSTDIGALM236 YPSYTFSGDV246 QLAQDDIDGI256 QAIYGRS |
|||||
|
|
||||||
| Click to View More Binding Site Information of This Target with Different Ligands | ||||||
| Different Human System Profiles of Target | Top |
|---|---|
|
Human Similarity Proteins
of target is determined by comparing the sequence similarity of all human proteins with the target based on BLAST. The similarity proteins for a target are defined as the proteins with E-value < 0.005 and outside the protein families of the target.
A target that has fewer human similarity proteins outside its family is commonly regarded to possess a greater capacity to avoid undesired interactions and thus increase the possibility of finding successful drugs
(Brief Bioinform, 21: 649-662, 2020).
Human Tissue Distribution
of target is determined from a proteomics study that quantified more than 12,000 genes across 32 normal human tissues. Tissue Specificity (TS) score was used to define the enrichment of target across tissues.
The distribution of targets among different tissues or organs need to be taken into consideration when assessing the target druggability, as it is generally accepted that the wider the target distribution, the greater the concern over potential adverse effects
(Nat Rev Drug Discov, 20: 64-81, 2021).
Human Pathway Affiliation
of target is determined by the life-essential pathways provided on KEGG database. The target-affiliated pathways were defined based on the following two criteria (a) the pathways of the studied target should be life-essential for both healthy individuals and patients, and (b) the studied target should occupy an upstream position in the pathways and therefore had the ability to regulate biological function.
Targets involved in a fewer pathways have greater likelihood to be successfully developed, while those associated with more human pathways increase the chance of undesirable interferences with other human processes
(Pharmacol Rev, 58: 259-279, 2006).
Biological Network Descriptors
of target is determined based on a human protein-protein interactions (PPI) network consisting of 9,309 proteins and 52,713 PPIs, which were with a high confidence score of ≥ 0.95 collected from STRING database.
The network properties of targets based on protein-protein interactions (PPIs) have been widely adopted for the assessment of target’s druggability. Proteins with high node degree tend to have a high impact on network function through multiple interactions, while proteins with high betweenness centrality are regarded to be central for communication in interaction networks and regulate the flow of signaling information
(Front Pharmacol, 9, 1245, 2018;
Curr Opin Struct Biol. 44:134-142, 2017).
Human Similarity Proteins
Human Tissue Distribution
Human Pathway Affiliation
Biological Network Descriptors
|
|
|
There is no similarity protein (E value < 0.005) for this target
|
|
Note:
If a protein has TS (tissue specficity) scores at least in one tissue >= 2.5, this protein is called tissue-enriched (including tissue-enriched-but-not-specific and tissue-specific). In the plots, the vertical lines are at thresholds 2.5 and 4.
|
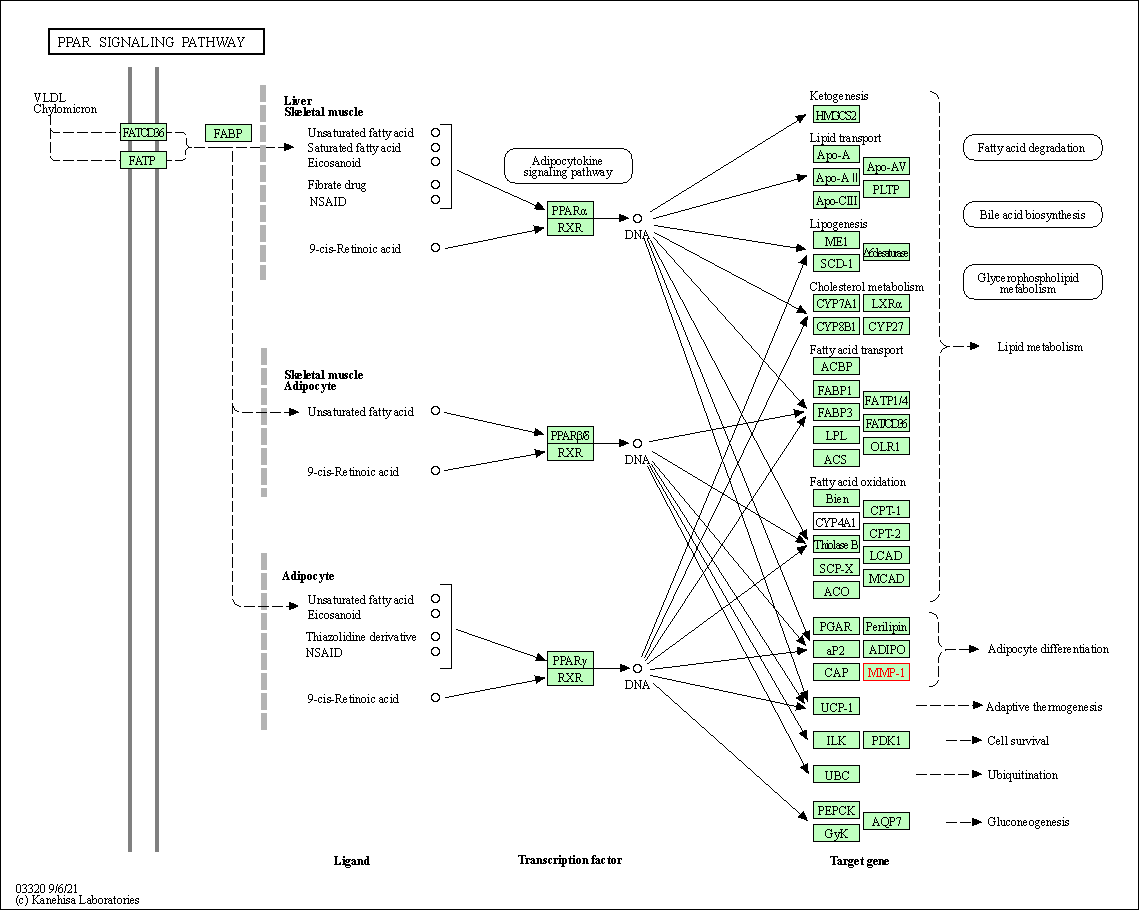
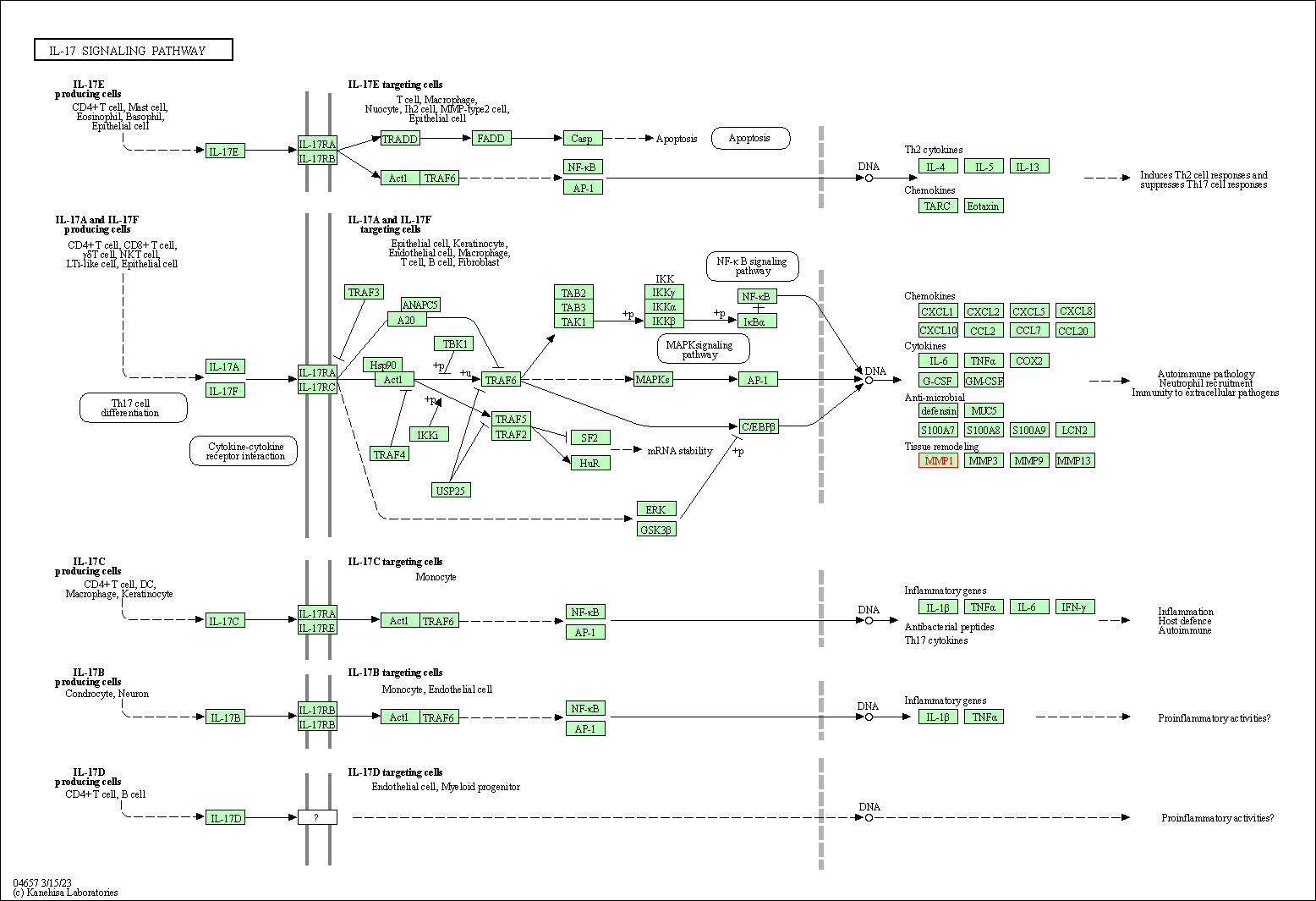
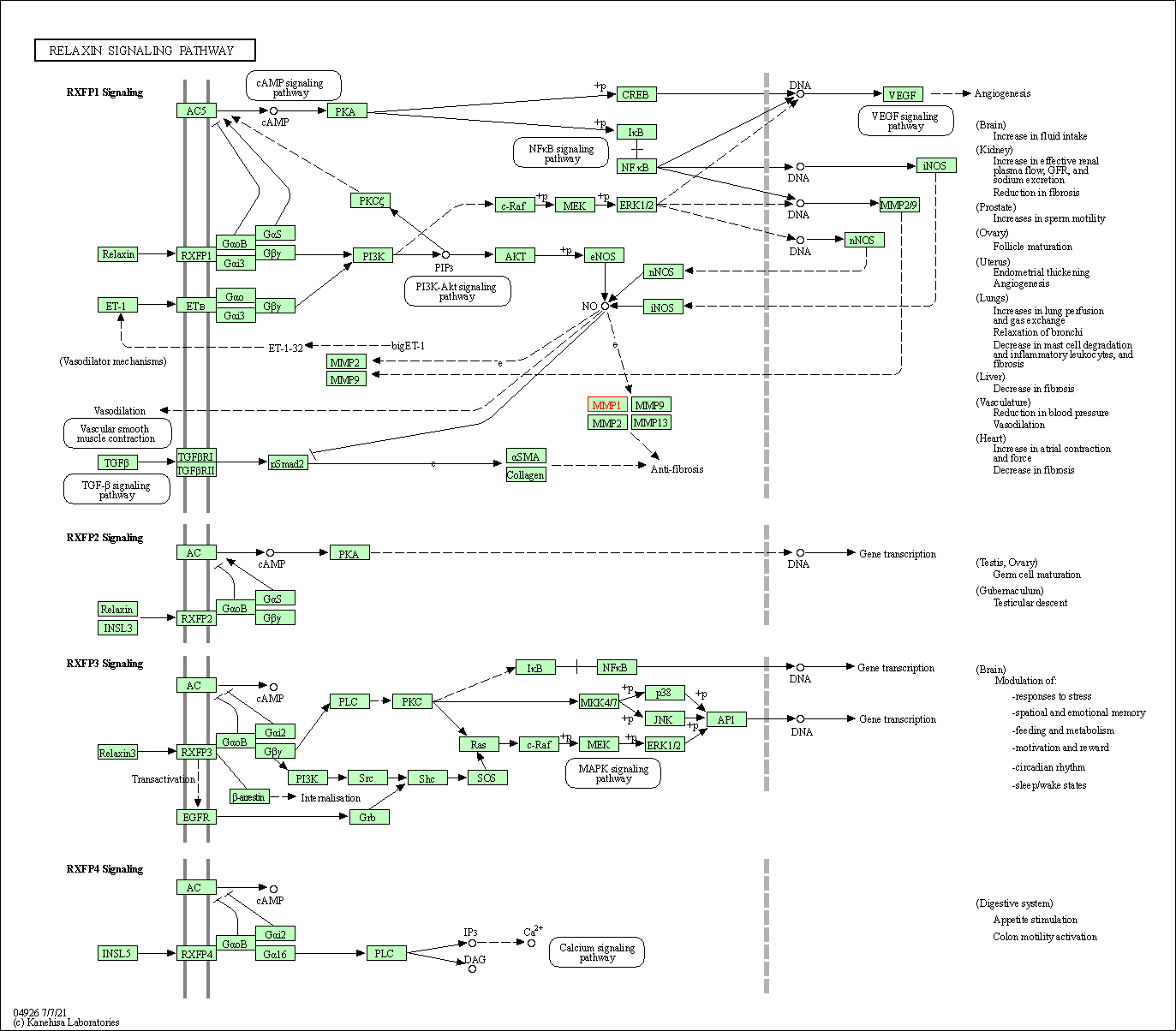
| KEGG Pathway | Pathway ID | Affiliated Target | Pathway Map |
|---|---|---|---|
| PPAR signaling pathway | hsa03320 | Affiliated Target |
|
| Class: Organismal Systems => Endocrine system | Pathway Hierarchy | ||
| IL-17 signaling pathway | hsa04657 | Affiliated Target |
|
| Class: Organismal Systems => Immune system | Pathway Hierarchy | ||
| Relaxin signaling pathway | hsa04926 | Affiliated Target |
|
| Class: Organismal Systems => Endocrine system | Pathway Hierarchy | ||
| Degree | 13 | Degree centrality | 1.40E-03 | Betweenness centrality | 3.18E-04 |
|---|---|---|---|---|---|
| Closeness centrality | 2.28E-01 | Radiality | 1.40E+01 | Clustering coefficient | 5.26E-01 |
| Neighborhood connectivity | 3.93E+01 | Topological coefficient | 1.33E-01 | Eccentricity | 12 |
| Download | Click to Download the Full PPI Network of This Target | ||||
| Chemical Structure based Activity Landscape of Target | Top |
|---|---|
| Drug Property Profile of Target | Top | |
|---|---|---|
| (1) Molecular Weight (mw) based Drug Clustering | (2) Octanol/Water Partition Coefficient (xlogp) based Drug Clustering | |
|
|
||
| (3) Hydrogen Bond Donor Count (hbonddonor) based Drug Clustering | (4) Hydrogen Bond Acceptor Count (hbondacc) based Drug Clustering | |
|
|
||
| (5) Rotatable Bond Count (rotbonds) based Drug Clustering | (6) Topological Polar Surface Area (polararea) based Drug Clustering | |
|
|
||
| "RO5" indicates the cutoff set by lipinski's rule of five; "D123AB" colored in GREEN denotes the no violation of any cutoff in lipinski's rule of five; "D123AB" colored in PURPLE refers to the violation of only one cutoff in lipinski's rule of five; "D123AB" colored in BLACK represents the violation of more than one cutoffs in lipinski's rule of five | ||
| Co-Targets | Top | |||||
|---|---|---|---|---|---|---|
| Co-Targets | ||||||
| Target Poor or Non Binders | Top | |||||
|---|---|---|---|---|---|---|
| Target Poor or Non Binders | ||||||
| Target Regulators | Top | |||||
|---|---|---|---|---|---|---|
| Target-regulating microRNAs | ||||||
| Target-regulating Transcription Factors | ||||||
| Target Profiles in Patients | Top | |||||
|---|---|---|---|---|---|---|
| Target Expression Profile (TEP) |
||||||
| Target-Related Models and Studies | Top | |||||
|---|---|---|---|---|---|---|
| Target Validation | ||||||
| Target QSAR Model | ||||||
| References | Top | |||||
|---|---|---|---|---|---|---|
| REF 1 | AG-3340 (Agouron Pharmaceuticals Inc). IDrugs. 2000 Mar;3(3):336-45. | |||||
| REF 2 | URL: http://www.guidetopharmacology.org Nucleic Acids Res. 2015 Oct 12. pii: gkv1037. The IUPHAR/BPS Guide to PHARMACOLOGY in 2016: towards curated quantitative interactions between 1300 protein targets and 6000 ligands. (Ligand id: 6505). | |||||
| REF 3 | Emerging therapies for neuropathic pain. Expert Opin Emerg Drugs. 2005 Feb;10(1):95-108. | |||||
| REF 4 | URL: http://www.guidetopharmacology.org Nucleic Acids Res. 2015 Oct 12. pii: gkv1037. The IUPHAR/BPS Guide to PHARMACOLOGY in 2016: towards curated quantitative interactions between 1300 protein targets and 6000 ligands. (Ligand id: 6466). | |||||
| REF 5 | Matrix metalloproteinase inhibition lowers mortality and brain injury in experimental pneumococcal meningitis. Infect Immun. 2014 Apr;82(4):1710-8. | |||||
| REF 6 | URL: http://www.guidetopharmacology.org Nucleic Acids Res. 2015 Oct 12. pii: gkv1037. The IUPHAR/BPS Guide to PHARMACOLOGY in 2016: towards curated quantitative interactions between 1300 protein targets and 6000 ligands. (Ligand id: 5220). | |||||
| REF 7 | Randomized phase III trial of marimastat versus placebo in patients with metastatic breast cancer who have responding or stable disease after first-line chemotherapy: Eastern Cooperative Oncology Group trial E2196. J Clin Oncol. 2004 Dec 1;22(23):4683-90. | |||||
| REF 8 | URL: http://www.guidetopharmacology.org Nucleic Acids Res. 2015 Oct 12. pii: gkv1037. The IUPHAR/BPS Guide to PHARMACOLOGY in 2016: towards curated quantitative interactions between 1300 protein targets and 6000 ligands. (Ligand id: 6482). | |||||
| REF 9 | ClinicalTrials.gov (NCT00095342) Study Evaluating TMI-005 in Active Rheumatoid Arthritis. U.S. National Institutes of Health. | |||||
| REF 10 | Phase 1/2 trial of BMS-275291 in patients with human immunodeficiency virus-related Kaposi sarcoma: a multicenter trial of the AIDS Malignancy Consortium. Cancer. 2008 Mar 1;112(5):1083-8. | |||||
| REF 11 | Trusted, scientifically sound profiles of drug programs, clinical trials, safety reports, and company deals, written by scientists. Springer. 2015. Adis Insight (drug id 800001387) | |||||
| REF 12 | Trusted, scientifically sound profiles of drug programs, clinical trials, safety reports, and company deals, written by scientists. Springer. 2015. Adis Insight (drug id 800010620) | |||||
| REF 13 | Agents in development for the treatment of diabetic nephropathy. Expert Opin Emerg Drugs. 2008 Sep;13(3):447-63. | |||||
| REF 14 | Trusted, scientifically sound profiles of drug programs, clinical trials, safety reports, and company deals, written by scientists. Springer. 2015. Adis Insight (drug id 800006361) | |||||
| REF 15 | A phase I and pharmacological study of the matrix metalloproteinase inhibitor BB-3644 in patients with solid tumours. Br J Cancer. 2004 Feb 23;90(4):800-4. | |||||
| REF 16 | Effects of the hydroxamic acid derivate Ro 31-4724 on the metabolism and morphology of interleukin-1-treated cartilage explants. Pharmacology. 1997 Aug;55(2):95-108. | |||||
| REF 17 | Trusted, scientifically sound profiles of drug programs, clinical trials, safety reports, and company deals, written by scientists. Springer. 2015. Adis Insight (drug id 800002350) | |||||
| REF 18 | Inhibition of gelatinase activity reduces neural injury in an ex vivo model of hypoxia-ischemia. Neuroscience. 2009 Jun 2;160(4):755-66. | |||||
| REF 19 | Delayed administration of a matrix metalloproteinase inhibitor limits progressive brain injury after hypoxia-ischemia in the neonatal rat. J Neuroinflammation. 2008 Aug 11;5:34. | |||||
| REF 20 | Pharmacoproteomics of a metalloproteinase hydroxamate inhibitor in breast cancer cells: dynamics of membrane type 1 matrix metalloproteinase-mediat... Mol Cell Biol. 2008 Aug;28(15):4896-914. | |||||
| REF 21 | Tumour microenvironment - opinion: validating matrix metalloproteinases as drug targets and anti-targets for cancer therapy. Nat Rev Cancer. 2006 Mar;6(3):227-39. | |||||
| REF 22 | Metalloelastase (MMP-12) induced inflammatory response in mice airways: effects of dexamethasone, rolipram and marimastat. Eur J Pharmacol. 2007 Mar 15;559(1):75-81. | |||||
| REF 23 | Matrix metalloproteinase-2 involvement in breast cancer progression: a mini-review. Med Sci Monit. 2009 Feb;15(2):RA32-40. | |||||
| REF 24 | Matrix metalloproteinase inhibition as a novel anticancer strategy: a review with special focus on batimastat and marimastat. Pharmacol Ther. 1997;75(1):69-75. | |||||
| REF 25 | Acetylenic TACE inhibitors. Part 3: Thiomorpholine sulfonamide hydroxamates. Bioorg Med Chem Lett. 2006 Mar 15;16(6):1605-9. | |||||
| REF 26 | Gelatinase inhibitors: a patent review (2011-2017).Expert Opin Ther Pat. 2018 Jan;28(1):31-46. | |||||
| REF 27 | Randomized phase II feasibility study of combining the matrix metalloproteinase inhibitor BMS-275291 with paclitaxel plus carboplatin in advanced non-small cell lung cancer. Lung Cancer. 2004 Dec;46(3):361-8. | |||||
| REF 28 | Randomized phase III study of matrix metalloproteinase inhibitor BMS-275291 in combination with paclitaxel and carboplatin in advanced non-small-cell lung cancer: National Cancer Institute of Canada-Clinical Trials Group Study BR.18. J Clin Oncol. 2005 Apr 20;23(12):2831-9. | |||||
| REF 29 | Introduction of the 4-(4-bromophenyl)benzenesulfonyl group to hydrazide analogs of Ilomastat leads to potent gelatinase B (MMP-9) inhibitors with i... Bioorg Med Chem. 2008 Sep 15;16(18):8745-59. | |||||
| REF 30 | Structure-based design of potent and selective inhibitors of collagenase-3 (MMP-13). Bioorg Med Chem Lett. 2005 Feb 15;15(4):1101-6. | |||||
| REF 31 | Broad spectrum matrix metalloproteinase inhibitors: an examination of succinamide hydroxamate inhibitors with P1 C alpha gem-disubstitution. Bioorg Med Chem Lett. 1998 Jun 16;8(12):1443-8. | |||||
| REF 32 | Inhibition of matrix metalloproteinases by N-carboxyalkyl peptides containing extended alkyl residues At P1', Bioorg. Med. Chem. Lett. 5(6):539-542 (1995). | |||||
| REF 33 | Receptor flexibility in the in silico screening of reagents in the S1' pocket of human collagenase. J Med Chem. 2004 May 20;47(11):2761-7. | |||||
| REF 34 | The asymmetric synthesis and in vitro characterization of succinyl mercaptoalcohol and mercaptoketone inhibitors of matrix metalloproteinases. Bioorg Med Chem Lett. 1998 May 19;8(10):1163-8. | |||||
| REF 35 | A potent, selective inhibitor of matrix metalloproteinase-3 for the topical treatment of chronic dermal ulcers. J Med Chem. 2003 Jul 31;46(16):3514-25. | |||||
| REF 36 | Synthesis and identification of conformationally constrained selective MMP inhibitors. Bioorg Med Chem Lett. 1999 Jul 5;9(13):1757-60. | |||||
| REF 37 | Discovery of a novel series of selective MMP inhibitors: identification of the gamma-sulfone-thiols. Bioorg Med Chem Lett. 1999 Apr 5;9(7):943-8. | |||||
| REF 38 | Hydroxamic acid derivatives as potent peptide deformylase inhibitors and antibacterial agents. J Med Chem. 2000 Jun 15;43(12):2324-31. | |||||
| REF 39 | Receptor flexibility in de novo ligand design and docking. J Med Chem. 2005 Oct 20;48(21):6585-96. | |||||
| REF 40 | The Protein Data Bank. Nucleic Acids Res. 2000 Jan 1;28(1):235-42. | |||||
| REF 41 | The discovery of a potent and selective lethal factor inhibitor for adjunct therapy of anthrax infection. Bioorg Med Chem Lett. 2006 Feb 15;16(4):964-8. | |||||
| REF 42 | Phosphoramidate peptide inhibitors of human skin fibroblast collagenase. J Med Chem. 1990 Jan;33(1):263-73. | |||||
| REF 43 | A cassette-dosing approach for improvement of oral bioavailability of dual TACE/MMP inhibitors. Bioorg Med Chem Lett. 2006 May 15;16(10):2632-6. | |||||
| REF 44 | 11,21-Bisphenyl-19-norpregnane derivatives are selective antiglucocorticoids, Bioorg. Med. Chem. Lett. 7(17):2299-2302 (1997). | |||||
| REF 45 | Design, synthesis, activity, and structure of a novel class of matrix metalloproteinase inhibitors containing a heterocyclic P2 P3 Bioorg. Med. Chem. Lett. 6(13):1541-1542 (1996). | |||||
| REF 46 | Synthesis and evaluation of succinoyl-caprolactam gamma-secretase inhibitors. Bioorg Med Chem Lett. 2006 May 1;16(9):2357-63. | |||||
| REF 47 | NMR solution structure of the catalytic fragment of human fibroblast collagenase complexed with a sulfonamide derivative of a hydroxamic acid compound. Biochemistry. 1999 Jun 1;38(22):7085-96. | |||||
| REF 48 | 1.56 A structure of mature truncated human fibroblast collagenase. Proteins. 1994 Jun;19(2):98-109. | |||||
If You Find Any Error in Data or Bug in Web Service, Please Kindly Report It to Dr. Zhou and Dr. Zhang.